



作者:药时代团队康明月整理
在国家以患者为中心、以临床价值为导向的政策指导下,越来越多的中国药企开始聚焦未满足的临床需求,而儿童用药就是其中之一。大量的常用药说明书并没有儿童用药信息,往往只写着儿童用量酌减或者遵医嘱,儿童用药剂量靠猜、分药靠掰是很普遍的现象,这也增加了儿童用药的风险。
7月28日,朗华制药副总裁毕明达博士受邀做客药时代第111期直播,为大家解读欧美儿科药物开发相关的法规和指导原则,以期帮助全球制药企业更好地推进儿童药物开发,以及探讨如何借鉴欧美法规助力国内儿童用药研发向国际接轨。
错过本期直播的小伙伴,点击「腾讯视频/bilibili」,观看视频回放,了解更多精彩内容!
全球儿科用药研发现状
根据以往儿科药物开发的经验教训,发现儿童群体和成人群体,在药动和剂量上是不同的。由于这种不同,药物在儿童群体中安全性和有效性的评估,是非常重要的,特别是PK方面的评估,但目前这种评估是很匮乏的。
为什么会引起这种评估的匮乏?总结来说,主要有以下原因:
第一种原因是,药物在儿童群体的测试不被鼓励。因为在儿童身体上测试药物,会有道德上的顾虑。此外,还有安全上的担忧,怕伤害到儿童。有的人还认为,在儿童身体上测试药物的责任远远大于在成人群体中测试,这也是一个因素。有些人认为儿童和成人只在体重方面不同,其他都相同。种种原因导致了药物在儿童群体中的测试不被鼓舞。
第二种原因是,药物在儿童群体中评估有内在的难度。这里毕明达博士列举了3个理由,儿科病人数量相对成人要少;儿童病来得及、但恢复的快,这种情况对药物公司来说不具有药物开发的吸引力;开发儿科药物需要较多的support体系,包括厂房、设备、实验室和相关的技能,这些系统相对成人来说比较匮乏。
但导致匮乏最重要的一点是,儿童药物开发的法规匮乏。如果没有法规、没有指导原则、不能要求药物公司开发儿科药,就改变不了现在儿科药物匮乏的现状。
PREA与BPCA的区别有哪些?
解决这种匮乏需要做出两种改变:一是观念上的改变,25年前的观念是为了保护儿童,我们不在儿童身体上测试药物,而现在的观念是我们要在儿童身体上测试,以保护他们;二是一定要拟定儿科的药物开发法规和指导原则。
以美国为例,第一个比较正式的儿童药物开发法规是1997年的FDA Modernization Act(FDAMA)。2002年,美国的参议院和众议院拟定了一种奖励性的法规,叫Best Pharmaceuticals for Children Act,简称为BPCA。它不是强制性的,该规定要求FDA可以邀请药物公司参与,但药物公司有权利决定参与还是不参与,也因此导致了法规的overall效果不是很明显。
继而,在2003年美国的参议院和众议院又拟定了一个新的法规叫Pediatric Research Equity Act,简称为PREA。这是一个强制性的法规,法规要求药物公司的某些药物需要对儿童群体进行安全性和有效性的评估。
BPCA和PREA这两个法规,每5年需要被参众两院评估一下,经过多年的数据调研,最后在2012年被列为永久的法规。调研表明,这两个法规对人类的健康,特别是儿童的健康非常有帮助。

(图:PREA与BPCA的对比)
除了以上提到的奖励性和强制性的不同外,第二点不同是,PREA主要针对成人指征,在儿童群体进行有效性和安全性的评估,而BPCA不只限于成人指征,可能会有一些其他指征,会邀请药物公司对额外的指征进行一些有效性和安全性的评估。
两者第三点不同在于对孤儿药的约束,PREA对孤儿药没有法律效力,无法要求孤儿药公司在儿童群体中进行安全性和有效性评估。而BPCA能邀请药物公司参与孤儿药在儿童群体安全性和有效性的评估,但是否参与由药物公司决定。
此外,2017年美国参众两院又拟定了一个新的儿童药物开发法规,叫RACE act。其主要要求药物公司的一些肿瘤药需要在儿童群体中进行有效性和安全性的评估。由于PREA对大部分肿瘤药基本没有法律效力,这一法规弥补了2003年PREA的漏洞。
PREA如何助力儿童药物商业化发展?
PREA在以下情况会起作用:一是药物新的指征申请上市时,或新的剂型申请上市时,或新的治疗方案、不同给药途径或创新药申请上市时,这些情况PREA都会要求药物公司在儿童群体中进行安全性和有效性评估。
PREA可以说是强制性要求药物公司的某些药在儿童群体中进行有效性和安全性评估,但同时也允许这些药物公司在条件允许的情况下申请豁免,这个豁免包括全部豁免和部分豁免。全部豁免指所有年龄段的儿童都不需要进行儿科药物开发或者评估。部分豁免只能申请一部分年龄段的儿科药物开发、评估及豁免,其他年龄段还需要进行评估。如果药物公司不符合申请豁免的条件,PREA也允许药物公司申请儿科药物开发延期。
Waivers and Deferrals鼓励药物公司与FDA在药物开发早期尽早沟通。但是因为早期的一些现有数据非常缺乏,所以FDA会最终决定是否给予豁免或给予延期,是在新药上市时,即NDA或者BLA申请阶段。
为了顺应PREA,药物公司首先需要填写全部的Pediatric Study Plan(PSP)。计划有12项,包括申请豁免、延期,儿科剂型的开发,临床前及各种可能的临床试验,选择哪个年龄段的儿童,时间点等。药物公司把PSP计划写好后,递交给FDA,经FDA审阅后,双方达成共识,PSP由此生效。

(图:Pediatric Study Plan的内容)
药企在递交PSP后,一般FDA会在90天工作日之内,给药物公司一个反馈,让他们进行评估及方案的修改。早期通过的PSP称为initial PSP(iPSP),因其通过仅是根据当时仅有的一些数据和信息,并不一定完善。所以,iPSP会随着后续项目的进展以及数据收集的情况进行修改。
如果某些公司不按照PREA法规开发儿科药物,可能导致其产品无法上市。由此可见遵守PREA法规的重要性。
BPCA如何助力儿童药物商业化发展?
PREA用PSP作为工具在FDA和药物公司之间进行沟通,而BPCA则是通过written request协调FDA与药物公司的沟通。written request是由FDA拟写的,然后再颁发给药物公司。这与PREA不同,PREA是由药物公司自己先撰写儿童药物开发方案,然后递交给FDA审阅。
FDA通过written request与药物公司达成共识后,药物公司如果愿意开发并能在规定的时间内完成,FDA会给予药物公司一个奖励:pediatric exclusivity,即给药物公司产品额外的6个月市场销售权延伸,且所有与drug moiety相关的专利都有6个月的延伸。这6个月通常会带来非常可观的收入,特别是对那些重磅药物。
欧洲儿童药物开发法规解读
欧洲儿童药物开发法规于2006年拟定,2007年生效。欧洲法规跟美国法规体系没有太大的区别,也是一个强制性法规与一个奖励性法规。但欧洲强制性法规比美国法规相对而言更严格一些。
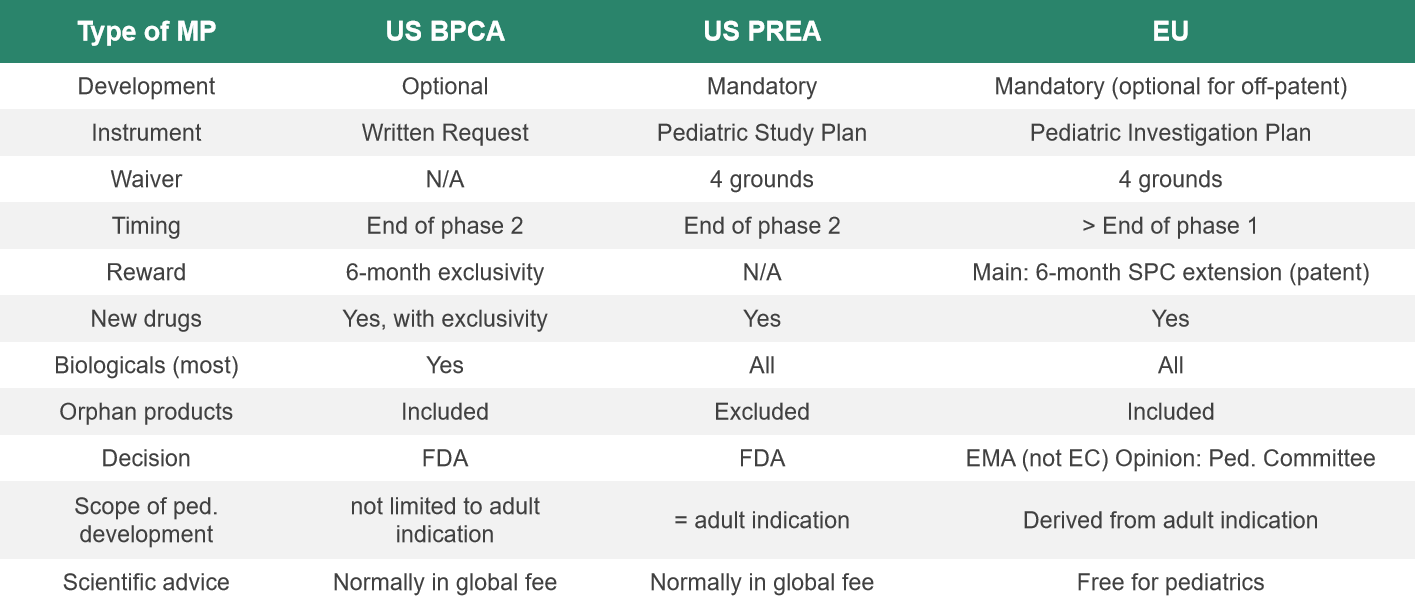
(图:欧美关键儿童药物开发法规的对比)
根据欧洲法规要求,药物公司的孤儿药需要在儿童群体中进行一些安全性和有效性评估。而在美国的法规中没有此类要求,PREA对孤儿药没有法律效力,BPCA可以邀请药物公司对孤儿药进行评估,由药物公司决定是否参与。其他方面,欧洲法规基本和美国法规相似。
欧洲法规同样也会给予奖励,大部分是6个月的市场销售权延伸和专利6个月的延伸。同时在欧洲孤儿药本身就拥有10年的市场销售期,要比美国多3年,且药物在做了儿科评估后,又可以得到2年额外的市场销售延伸。而在美国,如果按照BPCA标准对孤儿药进行评估,只能得到6个月延伸。所以与美国法规相比,欧洲法规更为严格,但激励幅度更大。
欧美儿童用药法规成效总结与启示
通过毕明达博士的深入解读,可以清晰地看到欧美儿童药物开发相关法规的发展历程和立法逻辑。美国在儿童药物开发方面,既有强制性法规PREA,也有奖励性法规BPCA,以及一个针对肿瘤药的补充法规RACE act,在监管方面可以说是张弛有度、收放自如,值得中国制药借鉴学习。欧洲法规跟美国相似,但在强制性法规方面更严格一些。从奖励方面看,欧洲的市场销售延伸时间更长,因此盈利的空间也更大。但无论是美国还是欧洲,都把儿童药物开发法规的拟定,作为改变儿科药物匮乏的关键。只有清晰明确的法规出台,才能规范和引导儿童药研发有序发展。期待中国制药在儿童药开发方面也能有更大的提高和改进,为更多的儿童患者带来福音。
小分子药物研发、生产一站式CDMO服务
朗华拥有一流的研发团队、充足的产能供应、先进的原料药和制剂技术平台、可靠的GMP和EHS管理体系及完善的IP保护体系等,可提供从原料药到制剂,从临床前到商业化供应,覆盖药物全生命周期的高效、灵活、高质量的一站式新药CDMO解决方案。

如需了解更多信息,可通过info@vivabiotech.com邮件与我们专家团队沟通。
Copyright © 维亚生物 All Rights Reserved. 沪ICP备19036061号